Researchers first discovered a new canine coronavirus in two Malaysian human patients infected with pneumonia in 2017-18. Another group of scientists isolated canine coronavirus, sequenced it, and published their findings in 2021.
A research team led by scientists from Cornell University and Temple University now found A pattern that appears at the end of the spike protein of canine coronavirus - the part of the virus that allows access to host cells: the virus changes from infecting the intestinal and respiratory system to the respiratory system of animal hosts that infect only human hosts.
The researchers found a terminal change - called the N-terminal - in a region of the molecule that was also detected in another coronavirus that spread from bats to humans and caused the common cold.
"This study identifies some molecular mechanisms for the transfer of hosts from canine coronavirus to new human hosts, which may also be important for the transmission of new human coronavirus that we did not know before," Professor Michael Stanhope said. Public and ecosystem health at the College of veterinary medicine. The first author, Jordan Zehr, is a doctoral student at the co-author Sergei kosakovsky pond laboratory. He is a professor of biology at the Institute of genomics and evolutionary medicine at Temple University.
In this study, the researchers used the most advanced molecular evolution tool developed by pond laboratory to assess how the pressure of natural selection affects the evolution of canine coronavirus.
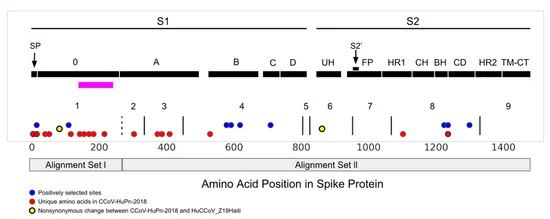
In humans, the main receptor that binds to alphacronavirus spike protein to enter human cells is called APN, but there are also auxiliary receptors. One of these common receptors is sialic acid, which exists in gastrointestinal cells of many mammals. The researchers found a spike protein region called O-domain at the N-terminal, which is famous for binding to sialic acid. When analyzing canine coronavirus found in Malaysian patients, some o-domains are changing in a unique way.
The study found that canine coronavirus found in Malaysian patients seems to be losing its O-domain - but not completely. "But its molecular evolutionary history suggests that the sialic acid binding region no longer does the same work," Stanhope said. The researchers found evidence of "loose evolution" and reduced pressure on natural selection, which contributed to the shift.
The researchers compared the transfer and loss of this O-domain with other related coronaviruses. A virus called porcine transmissible gastroenteritis virus (TGEV) infects pigs and causes respiratory and intestinal diseases. A variant of this porcine virus, called porcine respiratory coronavirus, is almost the same as TGEV, but it loses O-domain and is completely a respiratory pathogen. Similarly, a coronavirus known to cause the human common cold originated from the gastrointestinal virus of bats, lost O-domain and spread to human hosts as a respiratory virus.
"Therefore, this pattern seems to be repeated in the evolution of coronavirus, especially in the evolution of coronavirus associated with these sexual transitions. We initially transferred from gastrointestinal infection to another host, and now it is respiratory infection," Stanhope said.
In 2021, some people in Haiti also reported the same variant of canine coronavirus found in Malaysia. These people also had respiratory diseases. Stanhope said scientists need more research to understand whether the virus spontaneously transferred to humans in different parts of the world, or whether the coronavirus, which represents the eighth known human coronavirus, has been circulating in people for decades without being found.
The N-terminal domain of sars-cov2 of covid-19 has attracted more and more attention of researchers. This study provides additional reasons to focus on this specific region of molecules.